
Big Memory & Breaking the Bottleneck Behind SARS-CoV-2 Mutation Testing
Big Memory & Breaking the Bottleneck Behind SARS-CoV-2 Mutation Testing
COVID-19 offers countless lessons and reminders, not least of which is that viruses mutate seemingly anywhere and at any time. Rapid and thorough tracking can mean the difference between contained strains and spiking loss of life.
As of early February 2021, the CDC reported its “scaled-up” NS3 genomic surveillance system tracking up to 750 samples per week. But this is the first line of defense against new, potentially more virulent strains, and by now everyone understands the need for better testing, tracking, and analysis. With better, quicker genomic sequencing systems, labs could test more samples in less time.
Today’s need for better compute platforms able to improve genomic analysis is acute, and it will only remain so in the years to come. This won’t be our last virus emergency. The need for more and better genomic analysis platforms will remain acute long beyond SARS-CoV-2.
Genomics: Big Data and Tight Bottlenecks
In 2015, PLOS Biology estimated that, by 2025, YouTube would generate 1 to 2 exabytes (EB) of data annually. However, this number pales alongside genomics, which will continue with 7x year-over-year data storage growth and generate from 2EB to 40EB annually. The article estimates genomics data doubling every seven months. Given that the WHO notes how, with SARS-CoV-2 alone, “the data size of sequenced libraries can reach hundreds of gigabytes,” this estimate seems more than likely. Even Harvard University notes that “biomedical research groups around the world are producing more data than they can handle.”
Seeing how to remedy the problem requires examining where the genomics processing pinch points are. Conventional single-cell genomics analysis progresses through several established stages, beginning with the genomic sequencer. This yields large matrixes of data correlating individual genes and cells. From there, processing steps include quality control and noise removal, cell count normalization, filtering for genes with only desired features, reducing the number of factors to analyze, and clustering cells according to gene expression counts.
Machine learning figures heavily into working through these stages, as it can take automation of workloads to make processing such large datasets feasible. For example, one pipeline performs classification of cells by their genes. Once classification completes, the system can then progress to trajectory inference and establishing relationships between cells. This is a monumental task when datasets span from hundreds of gigabytes into terabytes.
Often, the servers tasked with processing these workloads are architecturally unable to maintain so much data in memory without resorting to one of two alternatives. In the case of a system cluster, the workload might be distributed across multiple machines, in which case coordinating the pieces of that dataset across systems introduces latency. Alternatively, the processing system can perform “disk swapping,” wherein parts of the workload can be moved from system memory into storage. Of course, it takes time to get that data out to storage and bring it back as needed, thereby introducing considerable processing delay.
Genomics analysis applications generally require intermediate output to storage between stages, further adding to potential delays. However, this intermediate output is often a vital part of the analysis process. Intermediate datasets can be iterated with changed parameters and then rerun at the preceding stage, effectively saying, “Let’s run that again, only with this little tweak made.” This iteration results in multiple workloads based on the same large dataset. Essentially, if the original dataset was 500GB, then two iterations of that would yield another 1TB or so of workload in addition to the original dataset. Since multiple researchers often want to run multiple iterations simultaneously, you can see how this process might very quickly exhaust available system memory. In some cases, such overtaxed servers will crash, sacrificing hours if not days of valuable processing time.
In short, memory is the primary bottleneck in genomics processing, which in turn can cascade into secondary bottlenecks in storage and/or networking. Matrix-heavy computations along with intermediate process stage writes to (and reloading from) storage create exceptional burdens in the genomics computation pipeline. Finishing just one pipeline with conventional system architecture can consume days to weeks.
The Big Memory Difference
After many years of development, persistent memory technology is finally ready to help burst this genomics workflow bottleneck. Persistent memory (PMem) is DRAM-like media that operates on the system memory bus yet is byte-addressable and non-volatile, like storage. On compatible server platforms, and with software written to take advantage of PMem’s functionality, applications can combine PMem with DRAM to create a significantly larger system memory pool. Additionally, some or all of that PMem resource can be sectioned off into non-volatile, extremely fast storage. In short, PMem offers lower price per gigabyte compared to DRAM, higher capacity points, and — when joined with DRAM and MemVerge’s Memory Machine software — faster-than-DRAM performance.
MemVerge uses PMem as part of its Memory Machine platform to enable ZeroIO™ memory snapshots. This is a MemVerge-specific technology in which intermediate data in system memory saves as a “snapshot” to non-volatile storage, except that storage just happens to reside in a partition of the PMem resources. Snapshot data never actually leaves the system’s memory modules. Thus, data iterations and reloads pull straight into PMem and DRAM from the exact same PMem. Transfer latency is almost non-existent.
If we examine the two operations of workload processing, computation and I/O, it becomes easy to see where so much of genomics analysis’s time burden comes from. Conventional architecture writes and reloads take thousands of seconds. With Memory Machine, that number drops to half of one second.
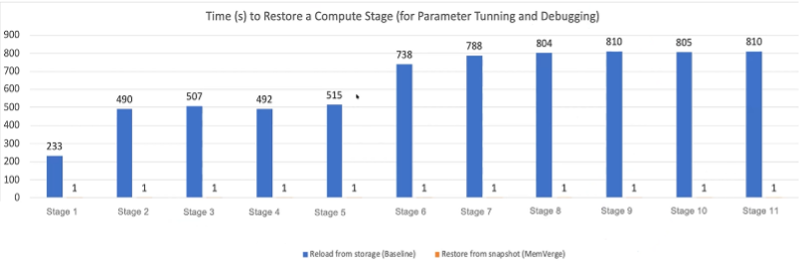
Naturally, there is still some overhead on running operations. I/O doesn’t vanish entirely. But the impact on total workload processing is incredible. MemVerge’s Big Memory testing reveals that conventional analysis of genomics-class workloads spend 61% of their runtime I/O (DRAM+SSD). This number drops to 3% with Memory Machine, effectively shaving off almost four hours of analysis processing time.


One customer reached out to MemVerge with what these results mean in the real world. Before adopting Memory Machine, the genomics lab could run 11 analysis iterations per day. After deploying Memory Machine, the lab could run 252 iterations daily — a nearly 23x improvement. Imagine what impact that could have on SARS-CoV-2 variant testing and tracking, never mind other genomics efforts throughout the world of precision medicine.
Moreover, the ability to maintain larger datasets in memory can yield higher classification accuracy. In 2017, Intel and Novartis teamed to create a multi-scale convolutional neural network (M-CNN) that would automatically locate objects of interest within images, extract features, and perform classification. The system downsampled a full-res image into smaller sizes for parallel analysis. Hardware limitations at the time required a batch size of eight, which required 16GB in memory. Researchers wanted batch sizes of 32 (64GB) and later 64 (80GB). Higher batch sizes turned out to yield higher classification accuracy and probability values that, in turn, enable better chemical treatment potency estimations. In other words, classification accuracy can be limited by memory size.[i]
Conclusion
SARS-CoV-2 is not our last deadly virus. The need for faster, better genomic analysis will only increase with time and the exponential growth of data. MemVerge’s Big Memory offers a breakthrough chance to keep pace with this growth by practically eliminating I/O and connectivity latencies caused by memory bottlenecking. Liberating memory capacity and performance restrictions will yield massive accelerating in the processing of genomic analysis pipelines, allowing researchers to learn faster and healthcare professionals to save more lives.
[i] Source: https://onlinexperiences.com/scripts/Server.nxp?LASCmd=AI:1;F:US!100&DisplayItem=EH242856&RandomValue=1573170006013; see approx. 42:00 to 46:00, requires login